Abstract
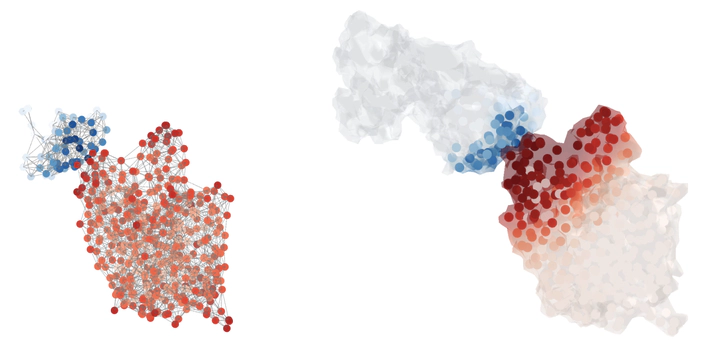
Identifying the binding sites of antibodies is essential for developing vaccines and synthetic antibodies. In this article, we investigate the optimal representation for predicting the binding sites in the two molecules and emphasize the importance of geometric information. Specifically, we compare different geometric deep learning methods applied to proteins’ inner (I-GEP) and outer (O-GEP) structures. We incorporate 3D coordinates and spectral geometric descriptors as input features to fully leverage the geometric information. Our research suggests that different geometrical representation information is useful for different tasks. Surface-based models are more efficient in predicting the binding of the epitope, while graph models are better in paratope prediction, both achieving significant performance improvements. Moreover, we analyze the impact of structural changes in antibodies and antigens resulting from conformational rearrangements or reconstruction errors. Through this investigation, we showcase the robustness of geometric deep learning methods and spectral geometric descriptors to such perturbations.
Marco Pegoraro
PostDoctoral Researcher, ISTA
I am a Ph.D. student in Geometric Deep Learning. My research activity is focused on spectral geometry processing applied to graph learning and computational biology.